Нормализация ДНК-микрочипов
Материал из MachineLearning.
м (→Парадигмы нормализации с точки зрения выбора опорного множества) |
|||
| (41 промежуточная версия не показана) | |||
| Строка 1: | Строка 1: | ||
| - | Нормализация - важный этап предобработки [[ДНК-микрочип]]ов, позволяющий сделать несколько рассматриваемых в эксперименте чипов пригодными к сравнению между собой. Основная цель анализа на этом этапе - исключить влияние систематических небиологических различий между микрочипами. Источников таких различий множество: вариации эффективности обратной транскрипции, маркировки красителями, гибридизации, физические различия между чипами | + | Нормализация - важный этап предобработки [[ДНК-микрочип]]ов, позволяющий сделать несколько рассматриваемых в эксперименте чипов пригодными к сравнению между собой. Основная цель анализа на этом этапе - исключить влияние систематических небиологических различий между микрочипами. Источников таких различий множество: вариации эффективности обратной транскрипции, маркировки красителями, гибридизации, физические различия между чипами, небольшие различия в концентрации реагентов, вариация лабораторных условий. |
| - | + | Показано, что выбор метода нормализации оказывает существенное влияние на результат анализа<ref name="norm">Hoffmann R, Seidl T, Dugas M. Profound effect of normalization on detection of differentially expressed genes in oligonucleotide microarray data analysis. Genome Biology. 2002;3(7):RESEARCH0033. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC126238/.</ref>. | |
| + | == Парадигмы нормализации с точки зрения выбора опорного множества == | ||
| + | Все методы нормализации в том или ином виде используют множество генов, экспрессия которых не должна меняться в условиях эксперимента, называемое опорным множеством (англ. reference set). Идея заключается в том, чтобы при поможи преобразований данных сделать распределение (среднее значение, дисперсию) интенсивности по этому множеству одинаковым по всем ДНК-микрочипам. | ||
| + | |||
| + | Один из подходов - использовать в качестве такого множества данные интенсивности по пробам, соответствующим так называемым генам домашнего хозяйства (англ. [http://en.wikipedia.org/wiki/Housekeeping_gene housekeeping genes]). Как правило, это гены, необходимые для поддержания базовых процессов жизнедеятельности клетки, и уровень их экспресии остаётся приблизительно постоянным в любых условиях. Гены, предлагаемые компанией Affymetrix в качестве генов домашнего хозяйства, часто использовались в качестве опорного множества для нормализации ДНК-микрочипов, однако, позднее было обнаружено, что при определённых экспериментальных условиях, например, при исследовании определённых патологий, экспрессия этих генов может значительно меняться<ref name="hcg">Ploner A, Miller LD, Hall P, Bergh J, Pawitan Y. Correlation test to assess low-level processing of high-density oligonucleotide microarray data. BMC bioinformatics. 2005;6(80). http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1084343/.</ref>. | ||
| + | |||
| + | Большинство используемых методов используют в качестве опорного множества множество всех генов, опираясь на следующие предположения: (1) экспрессия большинства генов на различных ДНК-микрочипов неизменна; (2) распределение изменения экспрессии близко к симетричному. Методы этого класса, как правило, неустойчивы к нарушению этих предположений; в то же время, на практике они проверяются нечасто. Кроме того, неясно. какую долю генов можно считать "большинством" - с биологической точки зрения этот вопрос не рассматривается, а со статистической необходимо требовать не менее 90% неизменных генов<ref name="lvs">Calza S, Valentini D, Pawitan Y. Normalization of oligonucleotide arrays based on the least-variant set of genes. BMC bioinformatics. 2008;9(140). http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2324100/.</ref>. | ||
| + | |||
| + | Существует также класс методов, строящих опорное множество непосредственно по данным конкретного эксперимента<ref name="lvs">Calza S, Valentini D, Pawitan Y. Normalization of oligonucleotide arrays based on the least-variant set of genes. BMC bioinformatics. 2008;9(140). http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2324100/.</ref><ref name="mlines">Li C, Wong WH. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biology. 2001;2(8):RESEARCH0032. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC55329/</ref>. Методы этого класса зависят от имеющихся данных и не используют предположений (1) и (2), либо позволяют непосредственно указать долю генов, экспрессия которых предполагается неизменной, из предположения (1). | ||
| + | |||
| + | Большинство методов нормализации, описываемых ниже, применимы при различных способах выбора опорного множества. | ||
| + | |||
| + | == Методы нормализации == | ||
=== Масштабирование === | === Масштабирование === | ||
Один из ДНК-микрочипов выбирается в качестве базового, затем все остальные масштабируются таким образом, чтобы их средняя интенсивность равнялась средней интенсивности базового (этот способ эквивалентен построению линейной регрессии каждого чипа на базовый и последующей нормализации при помощи регрессионной функции). | Один из ДНК-микрочипов выбирается в качестве базового, затем все остальные масштабируются таким образом, чтобы их средняя интенсивность равнялась средней интенсивности базового (этот способ эквивалентен построению линейной регрессии каждого чипа на базовый и последующей нормализации при помощи регрессионной функции). | ||
| - | Для большей устойчивости можно использовать усечённое среднее. Так, в стандартном программном обеспечении производителя микрочипов Affymetrix перед подсчётом среднего отбрасываются по 2% наибольших и наименьших значений интенсивности. Другая | + | Для большей устойчивости можно использовать усечённое среднее. Так, в стандартном программном обеспечении производителя микрочипов Affymetrix перед подсчётом среднего отбрасываются по 2% наибольших и наименьших значений интенсивности. Другая модификация — масштабирование к средней интенсивности не по всему базовому чипу, а по каждому подмножеству его проб, соответствующих одному гену. |
| - | Affymetrix предлагает использовать | + | Affymetrix предлагает использовать масштабирование на последнем этапе предобработки, применяя его непосредственно к матрицам экспрессии, однако, возможно и применение к матрицам интенсивности. |
| - | === Нелинейные методы=== | + | =====Схема выполнения масштабирования===== |
| + | Выбрать столбец <tex>j</tex> матрицы <tex>X</tex> в качестве базового. | ||
| + | Вычислить (усечённое) среднее <tex>\tilde{X}_j</tex> по столбцу <tex>j.</tex> | ||
| + | Для всех остальных столбцов матрицы <tex>X</tex>: вычислить (усечённое) среднее <tex>\tilde{X}_i</tex> по столбцу <tex>i</tex>; вычислить <tex>\beta_i=\tilde{X}_j/\tilde{X}_i</tex>; каждый элемент столбца <tex>i</tex> умножить на <tex>\beta_i.</tex> | ||
| + | |||
| + | === Нелинейные методы === | ||
| + | Предложено большое количество нелинейных способов нормализации данных, использующих различные настраиваемые функции, заменяющие линейную регрессию из предыдущего примера. Среди таких функций | ||
cross-validated splines<ref name="splines">Schadt EE, Li C, Ellis B. Feature extraction and normalization algorithms for high-density oligonucleotide gene expression array data. Journal of Cellular Biochemistry, Suppl. 2001;37:120-125. http://www.hsph.harvard.edu/~cli/pdf/Schadt_01.pdf</ref>, | cross-validated splines<ref name="splines">Schadt EE, Li C, Ellis B. Feature extraction and normalization algorithms for high-density oligonucleotide gene expression array data. Journal of Cellular Biochemistry, Suppl. 2001;37:120-125. http://www.hsph.harvard.edu/~cli/pdf/Schadt_01.pdf</ref>, | ||
running median lines<ref name="mlines">Li C, Wong WH. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biology. 2001;2(8):RESEARCH0032. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC55329/</ref>, | running median lines<ref name="mlines">Li C, Wong WH. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biology. 2001;2(8):RESEARCH0032. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC55329/</ref>, | ||
| - | loess smoothers<ref name="smoothers">Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185-193. http://www.ncbi.nlm.nih.gov/pubmed/12538238</ref> | + | loess smoothers<ref name="smoothers">Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185-193. http://www.ncbi.nlm.nih.gov/pubmed/12538238</ref>, |
| + | и т.д. В типичном случае нелинейная нормализация проводится по множеству рангово-инвариантных проб, то есть, проб, имеющих один и тот же ранг во всех микрочипах. | ||
| - | === | + | =====Схема выполнения нелинейной нормализации===== |
| - | === | + | Выбрать столбец <tex>j</tex> матрицы <tex>X</tex> в качестве базового. |
| - | [[ | + | Для всех столбцов <tex>i\neq\j</tex> матрицы настроить параметры нелинейной функции <tex>f</tex>, отображающей столбец <tex>i</tex> на столбец <tex>j</tex>. Пусть <tex>\hat{f}_i</tex> - полученное отображение. |
| - | + | Нормализованные значения в столбце <tex>j</tex> определяются как <tex>\hat{f}_i\left(X_j\right).</tex> | |
| - | <ref name=" | + | |
| + | === Квантильная нормализация === | ||
| + | [[Изображение:Quantile-quantile normalization.png|thumb|250px|Метод квантильной нормализации преобразует распределение интенсивностей.]] | ||
| + | Цель квантильной нормализации - сделать одинаковыми эмпирические распределения интенсивностей всех микрочипов. Для этого используется преобразование вида <tex>x'_i=F^{-1}\left[G\left(x_i\right)\right],</tex> где <tex>G</tex> - эмпирическое распределение интенсивностей каждого чипа, <tex>F</tex> - эмпирическое распределение интенсивностей усреднённого чипа. Можно модифицировать метод, оценивая <tex>F^{-1}</tex> и <tex>G</tex> более гладкими функциями. Однако, для данных большой размерности на практике достаточно и грубой оценки. | ||
| + | |||
| + | Приводимый метод квантильной нормализации - не единственный способ нормализации, основанный на квантилях. Описан метод, основанный на построении сплайнов по множеству квантилей<ref name="qsplines">Workman C, Jensen LJ, Jarmer H, et al. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome biology. 2002;3(9):RESEARCH0048. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC126873/</ref>, непараметрический метод присваивания одного и того же распределения каждому микрочипу<ref name="npar">Sidorov IA, Hosack DA, Gee D., et al. Oligonucleotide microarray data distribution and normalization. Information Sciences. 2002;146:67-73. http://www.ccrnp.ncifcrf.gov/~dimitrov/bibliogr/pdf/Sidorov2002InformSci.pdf</ref> и др. | ||
| + | |||
| + | =====Схема выполнения квантильной нормализации===== | ||
| + | Имея <tex>n</tex> микрочипов размерности <tex>p</tex>, построить матрицу <tex>X</tex> размерности <tex>p\times n</tex>, где в каждом столбце находятся значения интенсивности по каждому чипу. | ||
| + | Отсортировать все столбцы <tex>X</tex> независимо друг от друга, сформировав матрицу <tex>X_s.</tex> | ||
| + | Взять среднее по каждой строке матрицы <tex>X_s</tex> и создать <tex>X'_s</tex> - матрицу той же размерности, что и <tex>X</tex>, в каждой строке которой все значения равны среднему по строке <tex>X_s.</tex> | ||
| + | Получить матрицу <tex>X_n</tex>, переставив значения в столбцах <tex>X'_s</tex> в том порядке, в каком они стояли в исходной матрице <tex>X.</tex> | ||
| + | |||
| + | === Циклическая нормализация при помощи локальной регрессии (cyclic loess)=== | ||
| + | Данный метод основан на многократном применении [[Алгоритм LOWESS|локальной регрессии]] (англ. LOWESS, LOESS) к парам ДНК-микрочипов. Процедура перебирает все попарные комбинации микрочипов, повторяя процесс нормализации до достижения сходимости. Одним из её недостатков является необходимость <tex>O\left(n^2\right)</tex> применений локальной регресиии, хотя, обычно требуется всего один или два шага цикла. Значительно уменьшить время вычисления можно, используя подмножество данных для построения регрессионной зависимости. | ||
| + | |||
| + | Ниже приведён алгоритм циклической loess-нормализации. Индексы <tex>i</tex> и <tex>j</tex> соответствуют номерам микрочипов, а индекс <tex>k</tex> - номеру пробы или подмножества проб, соответствующих одному гену. | ||
| + | =====Схема выполнения циклической loess-нормализации===== | ||
| + | Сформировать матрицу <tex>X</tex> размерности <tex>p\times n,</tex> где столбцы соответствуют различным микрочипам, а строки - пробам или подмножествам проб. | ||
| + | Выполнить логарифмирование данных: <tex>X\leftarrow \log X.</tex> | ||
| + | Повторять | ||
| + | Цикл по <tex>i</tex> от <tex>1</tex> до <tex>n-1</tex> | ||
| + | Цикл по <tex>j</tex> от <tex>i+1</tex> до <tex>n</tex> | ||
| + | Цикл по <tex>k</tex> от <tex>1</tex> до <tex>p</tex> | ||
| + | Вычислить <tex>M_k=x_{ki}-x{kj}</tex> и <tex>A_k=\frac{1}{2}\left(x_{ki}+x{kj}\right)</tex> | ||
| + | Конец цикла по <tex>k</tex> | ||
| + | Построить локальную регрессию <tex>M</tex> на <tex>A</tex>, обозначим полученную зависимость <tex>\hat{f}.</tex> | ||
| + | Цикл по <tex>k</tex> от <tex>1</tex> до <tex>p</tex> | ||
| + | <tex>\hat{M}_k=\hat{f}\left(A_k\right)</tex> | ||
| + | <tex>a_k=\left(M_k-\hat{M}_k\right)/n</tex> | ||
| + | <tex>x_{ki}=x_{ki}+a_{k}</tex> и <tex>x_{kj}=x_{kj}-a_{k}</tex> | ||
| + | Конец цикла по <tex>k</tex> | ||
| + | Конец цикла по <tex>j</tex> | ||
| + | Конец цикла по <tex>i</tex> | ||
| + | до достижения сходимости или максимального числа итераций. | ||
| + | Выполнить преобразование к исходной шкале: <tex>X\leftarrow \exp\left(X\right).</tex> | ||
| + | |||
| + | == Литература == | ||
| + | Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S (Eds.). Bioinformatics and Computational Biology Solutions Using R and Bioconductor. (2005) 473 p. ISBN: 978-0-387-25146-2. | ||
== Примечания == | == Примечания == | ||
<references /> | <references /> | ||
| - | |||
[[Категория:Биоинформатика]] | [[Категория:Биоинформатика]] | ||
| - | |||
Текущая версия
Нормализация - важный этап предобработки ДНК-микрочипов, позволяющий сделать несколько рассматриваемых в эксперименте чипов пригодными к сравнению между собой. Основная цель анализа на этом этапе - исключить влияние систематических небиологических различий между микрочипами. Источников таких различий множество: вариации эффективности обратной транскрипции, маркировки красителями, гибридизации, физические различия между чипами, небольшие различия в концентрации реагентов, вариация лабораторных условий.
Показано, что выбор метода нормализации оказывает существенное влияние на результат анализа[1].
Содержание |
Парадигмы нормализации с точки зрения выбора опорного множества
Все методы нормализации в том или ином виде используют множество генов, экспрессия которых не должна меняться в условиях эксперимента, называемое опорным множеством (англ. reference set). Идея заключается в том, чтобы при поможи преобразований данных сделать распределение (среднее значение, дисперсию) интенсивности по этому множеству одинаковым по всем ДНК-микрочипам.
Один из подходов - использовать в качестве такого множества данные интенсивности по пробам, соответствующим так называемым генам домашнего хозяйства (англ. housekeeping genes). Как правило, это гены, необходимые для поддержания базовых процессов жизнедеятельности клетки, и уровень их экспресии остаётся приблизительно постоянным в любых условиях. Гены, предлагаемые компанией Affymetrix в качестве генов домашнего хозяйства, часто использовались в качестве опорного множества для нормализации ДНК-микрочипов, однако, позднее было обнаружено, что при определённых экспериментальных условиях, например, при исследовании определённых патологий, экспрессия этих генов может значительно меняться[1].
Большинство используемых методов используют в качестве опорного множества множество всех генов, опираясь на следующие предположения: (1) экспрессия большинства генов на различных ДНК-микрочипов неизменна; (2) распределение изменения экспрессии близко к симетричному. Методы этого класса, как правило, неустойчивы к нарушению этих предположений; в то же время, на практике они проверяются нечасто. Кроме того, неясно. какую долю генов можно считать "большинством" - с биологической точки зрения этот вопрос не рассматривается, а со статистической необходимо требовать не менее 90% неизменных генов[1].
Существует также класс методов, строящих опорное множество непосредственно по данным конкретного эксперимента[1][1]. Методы этого класса зависят от имеющихся данных и не используют предположений (1) и (2), либо позволяют непосредственно указать долю генов, экспрессия которых предполагается неизменной, из предположения (1).
Большинство методов нормализации, описываемых ниже, применимы при различных способах выбора опорного множества.
Методы нормализации
Масштабирование
Один из ДНК-микрочипов выбирается в качестве базового, затем все остальные масштабируются таким образом, чтобы их средняя интенсивность равнялась средней интенсивности базового (этот способ эквивалентен построению линейной регрессии каждого чипа на базовый и последующей нормализации при помощи регрессионной функции).
Для большей устойчивости можно использовать усечённое среднее. Так, в стандартном программном обеспечении производителя микрочипов Affymetrix перед подсчётом среднего отбрасываются по 2% наибольших и наименьших значений интенсивности. Другая модификация — масштабирование к средней интенсивности не по всему базовому чипу, а по каждому подмножеству его проб, соответствующих одному гену.
Affymetrix предлагает использовать масштабирование на последнем этапе предобработки, применяя его непосредственно к матрицам экспрессии, однако, возможно и применение к матрицам интенсивности.
Схема выполнения масштабирования
Выбрать столбецматрицы
в качестве базового. Вычислить (усечённое) среднее
по столбцу
Для всех остальных столбцов матрицы
по столбцу
; вычислить
; каждый элемент столбца
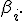
Нелинейные методы
Предложено большое количество нелинейных способов нормализации данных, использующих различные настраиваемые функции, заменяющие линейную регрессию из предыдущего примера. Среди таких функций cross-validated splines[1], running median lines[1], loess smoothers[1], и т.д. В типичном случае нелинейная нормализация проводится по множеству рангово-инвариантных проб, то есть, проб, имеющих один и тот же ранг во всех микрочипах.
Схема выполнения нелинейной нормализации
Выбрать столбецматрицы настроить параметры нелинейной функции
, отображающей столбец
- полученное отображение. Нормализованные значения в столбце
.)
Квантильная нормализация

Цель квантильной нормализации - сделать одинаковыми эмпирические распределения интенсивностей всех микрочипов. Для этого используется преобразование вида где
- эмпирическое распределение интенсивностей каждого чипа,
- эмпирическое распределение интенсивностей усреднённого чипа. Можно модифицировать метод, оценивая
и
более гладкими функциями. Однако, для данных большой размерности на практике достаточно и грубой оценки.
Приводимый метод квантильной нормализации - не единственный способ нормализации, основанный на квантилях. Описан метод, основанный на построении сплайнов по множеству квантилей[1], непараметрический метод присваивания одного и того же распределения каждому микрочипу[1] и др.
Схема выполнения квантильной нормализации
Имеямикрочипов размерности
, построить матрицу
, где в каждом столбце находятся значения интенсивности по каждому чипу. Отсортировать все столбцы
Взять среднее по каждой строке матрицы
и создать
- матрицу той же размерности, что и
, переставив значения в столбцах
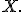
Циклическая нормализация при помощи локальной регрессии (cyclic loess)
Данный метод основан на многократном применении локальной регрессии (англ. LOWESS, LOESS) к парам ДНК-микрочипов. Процедура перебирает все попарные комбинации микрочипов, повторяя процесс нормализации до достижения сходимости. Одним из её недостатков является необходимость применений локальной регресиии, хотя, обычно требуется всего один или два шага цикла. Значительно уменьшить время вычисления можно, используя подмножество данных для построения регрессионной зависимости.
Ниже приведён алгоритм циклической loess-нормализации. Индексы и
соответствуют номерам микрочипов, а индекс
- номеру пробы или подмножества проб, соответствующих одному гену.
Схема выполнения циклической loess-нормализации
Сформировать матрицугде столбцы соответствуют различным микрочипам, а строки - пробам или подмножествам проб. Выполнить логарифмирование данных:
Повторять Цикл по
до
Цикл по
до
от
и
Конец цикла по
на
, обозначим полученную зависимость
Цикл по
)
/n)
и
Конец цикла по
.)
Литература
Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S (Eds.). Bioinformatics and Computational Biology Solutions Using R and Bioconductor. (2005) 473 p. ISBN: 978-0-387-25146-2.
